Computer-Aided Drug Design of Novel Pyrimidine Derivatives Targeting S-Phase Kinase Associated Protein-2: Molecular Docking and ADMET Evaluation
DOI:
https://doi.org/10.54117/jcddms.v4i1.59Keywords:
Computer-Aided Drug Design (CADD), SKP2, Pyrimidine Derivatives, Molecular Docking, ADMET, Anticancer Agents, 2ASS, Ubiquitin-Proteasome SystemAbstract
S-Phase Kinase Associated Protein-2 (SKP2), an F-box protein component of the Skp1-Cul1-F-box (SCF) ubiquitin ligase complex, is an oncoprotein frequently overexpressed in various human cancers. It facilitates the targeted ubiquitination and proteasomal degradation of key tumour suppressors, most notably p27Kip1, thereby promoting uncontrolled cellular proliferation and tumorigenesis. Consequently, SKP2 has emerged as a highly promising and validated molecular target for anticancer drug discovery.
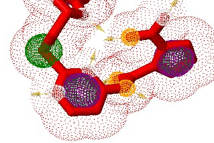
This research aimed to design novel pyrimidine-based derivatives as potent and selective inhibitors of SKP2 (PDB ID: 2ASS) using an integrated computational drug design approach. A focused library of novel pyrimidine analogues was designed based on the structural insights of the native ligand and known SKP2 inhibitors. Molecular docking studies were performed to investigate the binding modes, affinity, and molecular interactions (hydrogen bonding, hydrophobic, and π-π stacking) between the designed ligands and the active site of the SKP2 protein.
The docking results revealed that several designed compounds exhibited superior binding affinity compared to the reference ligand, and formed critical interactions with key residues in the SKP2 binding pocket. The top-ranking ligands were subsequently subjected to comprehensive in silico ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction to evaluate their drug-likeness and pharmacokinetic properties, including gastrointestinal absorption, blood-brain barrier penetration, cytochrome P450 inhibition, and potential hepatotoxicity.
The results demonstrate that the newly designed pyrimidine derivative, 6-((1H-pyrrole-2-carboxyl)oxy)2-(((2,3-dihydro-1H-benzo[d]imidazole-2-yl)methyl)thio)pyrimidin-4-carboxylic acid, possesses strong potential to inhibit SKP2 effectively and exhibits favourable ADMET properties with adherence to Lipinski’s Rule of Five, indicating promising potential as an orally bioavailable therapeutic agent. This study provides a robust foundation for the development of novel SKP2 inhibitors, identifying specific lead compounds worthy of further synthesis and experimental validation through in vitro and in vivo anticancer assays.
Downloads
Published
How to Cite
Issue
Section
License
Copyright (c) 2026 Anil Kumar Sharma, Min Prasad Subedi, P. Rashmi, Viswanath Sekar, Sharmila A. Gote, Kamlesh Yadav, Gowthami, A. Narendra

This work is licensed under a Creative Commons Attribution 4.0 International License.
